载体构建的一些心得
载体构建是分子生物学研究常用的手段之一。主要包括已有载体多克隆位点MCS的改造和已有载体启动子、增强子、筛选标记等功能元件的改造。载体构建完成后可利用PCR原理进行测序验证。今天小编整理了了一些关于载体构建的一些心得,供大家参考!

![]()
1、前期准备工作
这里推荐大家两个工具,一个都知道,PRIMER5.0。另一个工具极强大,叫lasergene,包括设计引物到构建图谱一应俱全,图谱非常漂亮,而且分析酶切位点等等。
2、PCR
如果没有现成的质粒可供酶切,PCR是理想也是方便的策略。
如何设计引物?
首先,看懂质粒图谱!
拿大家比较熟悉的PEGFP-C1和PEGFP-N1做例子。想用N1质粒,设计引物就得把下游引物上的中止密码子去掉,不要辛辛苦苦的一路做下来结果根本不表达融合蛋白,你就死了;C1质粒,注意frame,要是移码了,你也死了。而且PEGFP系列有1.2.3,要弄清楚别弄窜了。一句话,要看懂你的图谱!再多一句,PEGFP的XBA1和Bcl1位点不能用。
注意在设计酶切位点的时候要加保护碱基(大家要用T载体就当我没说)。酶切位点设计也有一定的讲究,我的原则是,能用粘端就用粘端,实在不能用的就用只好用一些常用的平端酶,如ECORV和SNAB1之类,要是以后还有别的用处就多加点酶切位点,我曾一口气在引物上加了五个酶切位点以防以后要用到,注意计算TM的时候要减去这些不match的序列,或者选用touch-up策略。
除了酶切位点,还要注意KOZAK序列的问题,很多质粒没有提供KOZAK序列,这要在设计的时候直接在引物上加好。
GENE OVERLAP也比较有用,比方说加个FLAG片段,HIS片段,2A序列之类的,直接设计三引物OVERLAP一下就可以,省得还要再多构建一步,这些都是设计引物时候就要考虑好的。
引物长一点不要紧(我喜欢两步法了),尤其是对GC比高的序列,有时候引物不长PCR根本不出结果,注意如果GC比较高,这个时候GENE OVERLAP就不要做了,很麻烦,克隆很难挑。
没有合适的酶切位点?
很简单,用同尾酶策略,比方说Bgl2和BamH1,Nhe1和spe1,Xho1和Sal1(注意连上了切不下来),实在不行就平端吧,只要不超过4KB,平端酶连接也不那么难。
如何选择Taq酶?
选择Taq酶也很关键,首先,高保真(High fidelity)这是必须的,我在国内常用LA KIT,可这边***忑贵,只好用(美)国货。常用的PFU,PHUSION和PFX。PFX50是invitrogen公司的,一度曾卖到脱销,保真性应该是目前高的,是普通Taq的50倍,对一般基因绝无问题(我同事4.5K PCR产物,居然一个突变都没有),但是对比较难PCR的高GC或者位点比较难做PCR的基因(基因组为模板)就明显不行了;PFU(安捷伦)保真性没有PFX50高,但是能力很强,略贵点;phusion是NEB公司的,不贵,但是这个酶很怪异,每次用都要把annealing温度调高好几度,否则就出杂带,个人觉得NEB内切酶绝对是NO.1,但是Taq就不如Invitrogen了,如果实验室有钱,直接买invitrogen的spuermix,什么都混好了,连ddH2o都搞定,只要直接向里加模版和引物就OK,每次我要拿漂亮的结果都用supermix。
还是那句话,读说明书。同是高保真Taq酶,iproof 要求98度起始1min,pfx就要求95起始2min ,延伸有的是72,有的是68,有的要加1uL,有的加0.5,有的TM要低五度,有的高三度,这些必须要读说明书,读且仔细读,这是拿到任何试剂都要做的第一步。
如果基因太难PCR,怎么办?
首先,DMSO是好物,好到甚至FISHER的Phusion就直接写上了DMSO这项,注意3%-6%,太高Taq酶活性就不行了。如果GC太高而且片段过长的话,DMSO也不行,GC低的不推荐。
我做个过一个2.8k,GC比高达92%的基因PCR,一共做了两周,从保真性强的PFX50到普通的promega 2xGO Taq都试过,什么DMSO,甘油,Biorad的iproof with high GC Buffer, NEB one Taq 2x Taq High GC(还带Enhancer的),TAKARA 的LA都试过,都不行,要么不出带,要么就是乱七八糟的带一起来,头晕脑涨的我都打算抹平策略了,后来从别的实验室弄来一个clontech的2 GC rich kit,一次搞定!强烈推荐这个KIT,太牛了,在别家都缴械的时候,它一锤定音,不过价格也比别家都牛,10次反应就130刀,其实实验室大可以备一个,就是防备超高GC又长的片段。不过这个KIT非高保真,送了三个克隆测序,各在不同部位有突变,于是我就从A克隆切一段接到B克隆上,又从C克隆切一段接B克隆上。注意的问题是GC比太高测序也很困难,正常GC能测1K左右,到了High GC就能测个五六百,这时要多准备些引物。
PCR产物的纯化
如果带很单一,又强,直接PCR产物纯化就可以,如果有杂带,但目的带也很强,跑胶,目的带切胶回收。回收这里也有个瓶颈,就是回收率的问题,我试过很多家的KIT,promega的GEL回收试剂盒效率和价格都很合适,推荐这个。
关于T载体,我在国内的时候是必用的,到了美帝反倒一次没用过,这边比较流行直接用PCR产物切,就是回收完了直接切上,回收后然后连接,这又回到了开始设计,要加保护碱基。这个策略好处就是免除了T载体这步,直接插入目的载体,存在的问题就是处于盲做状态,还要加保护碱基。
3. 酶切
原则一向是:尽量避免PCR,能酶切的多酶切,实在没办法才去PCR。
这里强烈推荐NEB的内切酶,还记得在有一次用别家的酶切过夜,结果质粒都被切碎了(当然当时质粒浓度也不高),而用NEB的酶,切个七十二小时仍旧一切OK,尤其现在NEB还推出了HF的内切酶,没有星活性而且统一都用Buffer4,很好用,在国内我都用TAKARA的酶,基本上还可以。
注意要读说明书,选用什么buffer,多少度,用不用加BSA,这些都要仔细读。
酶切的起始量,PCR产物没甚么可说的,全都切上,如果是从质粒上切片段,要根据你片段大小,片段适中,比如说7kb质粒,有2KB是目的片段,这时切个3-4ug就足够了,如果只有0.2KB,那好多切点,6ug-7ug 内切酶也没问题。
4. 连接
常用的是NEB的T4和Promega的T4,都很好用,但是注意Buffer里有ATP,反复冻融ATP失活得很快,这时有两种办法,一种是象我们chair实验室,每次连接都统统朝里面加1uL ATP;另一种是我们实验室的策略,拿到buffer就分装,10uL/管,一次性使用,哪怕就用1uL,剩下的直接扔掉。
连接重要的注意事项,1,载体总量,2.载体和插入片段比例,3.载体和插入片段质量。
我在回收之后都要测载体浓度,一般来说,载体浓度在20-100ng/uL很好,太低的话碰撞几率低,太高的话又会产生很多非目的克隆,总量从50-100ng就可以,太低失败几率很高,太高克隆太多,挑克隆会很麻烦,反应总体积也有讲究,体积太大载体和目的片段碰撞几率太低,而且对感受态细胞也是个挑战,通用10-15uL,我试过25uL没有问题,40uL就不行了。
载体和插入片段比例一般是1:3,如果很难连接,比如说平端连接,要适当提高载体浓度,同时加大比例1:10,我试过一端平一端粘连接,4kb片段,4KB载体,连接成功率相当高,10个克隆里8个都是阳性的,但是7KB片段,5KB载体的平端连接就连试两周都失败,后换策略分两段先后克隆进去的,这也給了我很深教训,隔了一阵又做类似的实验,这回我干脆连试都没试,继续分成两段分别连进去的。
3片段连接同理,如果片段不太大,比方说小于3K,3片段连接成功几率很高,但是如果你片段太大,就不行了,我试过4K载体,4K片段A,2K片段B连接,失败四五次,后还是绕路走了。
5. 转化
这个简单遵循基本的准则就行,话说实验室原来从sigma买感受态(competent cell),后来发现还是自己做的效率高,尤其在使用XL-10细菌的时比DH5a效率要高,需要的话我可以发protocol上来。要注意的是LB必须无菌而且没有抗生素,实验室原来有个volunteer,做一次失败一次,我就奇怪了,后来才发现他用的居然是加了KANA的LB,直接晕过去了。还有就是氯化钙的问题。氯化钙吸水性很强,吸水后就完蛋了,我一般把它放在干燥罐里(反正我也不知道啥名字,就那种玻璃罐,干燥用的),大家要不放心,用之前可以把氯化钙放在烘箱里烤一烤。
6. 鉴定
普通实验做酶切鉴定,阳性率非常高,大多数情况下四个中起码有三个是正确的,很多时候都是百分百正确,这里推荐一种叫fast digest的酶,切个十几分钟跑胶就行了。
如果找不到合适的酶切位点或者有其他问题,比方说有段时间我做个非常新潮的实验,初始步骤的虽然是蓝白筛选,但是白斑也大部分都不对,没办法鉴定就用菌液PCR,夸张的一次是五十个白斑里面居然就一个阳性的(没办法,该实验特点),这要提质粒可提不起。菌液PCR也有讲究,很多人做菌液PCR鉴定假阳性特多,为甚么?因为你PCR引物选的都在载体上,注意,引物必须分别在载体和插入片段上才准,PCR方法鉴定是极其准确的,我做过数百次菌液PCR,只要PCR条件正确,PCR和酶切结果绝对百分之百对得上。PCR鉴定做起来也很简单,拿个2mL tube,装0.5mL 加入相应抗生素的LB,摇个三小时左后取1uL做模板就行了,楼下有个实验室的POSTDOC特喜欢做直接的菌落PCR,我没试过,效果怎样不好说。
还要多说一句,要注意质粒是低拷贝还是高拷贝,普通的质粒一般4mL摇过夜,1.5mL就能提到500ng/uL 总体积40uL的质粒(根据试剂盒不同,高提过800ng/uL,也试过200ng/ul,都正常)。如果是低拷贝质粒,象PET系列和pshuttle系列,都是低拷贝,能拿到100ng/uL就已经很好了,这时候需要4ML菌当2ML提,但是要命的还是Adeasy 质粒(用来做腺病毒的),这个质粒超过30KB,普通KIT回收效率低到忍无可忍,一定要用特定的试剂盒才行,还是那句话,这些都要读说明书。
7. 测序
拿到测序结果时候,不仅要核对具体的序列,而且要看测序图,这很重要,因为有时候光看结果对,实际上图显示的不对,或者虽然结果不对,但是图上出现的峰值本身就很怪异不可信,出现突变也不怕,有很大可能性是简并密码子,还要重点检查的地方就是“接头”的地方,因为偶尔引物会出错,比方说少个碱基什么的,还有时候酶切之后连接也会丢一或者俩碱基什么的(这些问题我都碰到过),如果不仔细检查到了后期悔之无及。
以上就是小编整理的一些科研朋友们对于载体构建的一些心得,如果想要了解更多实验技术内容可以到本站发布自己遇到的实验技术难题,与科研朋友们一起探讨心得!
 打开微信“扫一扫”,打开网页后点击屏幕右上角分享按钮
打开微信“扫一扫”,打开网页后点击屏幕右上角分享按钮

-
论文打印要求是什么,单面还是双面? 132933
-
 ieee论文什么水平,含金量如何? 71704
ieee论文什么水平,含金量如何? 71704
-
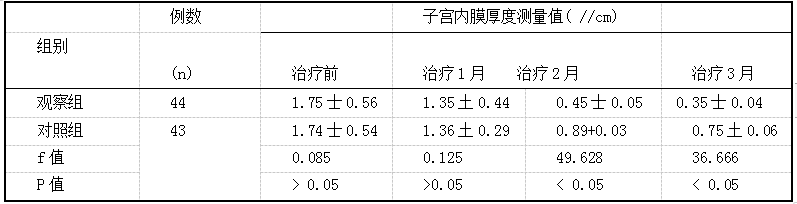 左炔诺孕酮不同给药途径对患者子宫内膜增生症的临床疗效评价 2019.10.30 15:05
左炔诺孕酮不同给药途径对患者子宫内膜增生症的临床疗效评价 2019.10.30 15:05 -
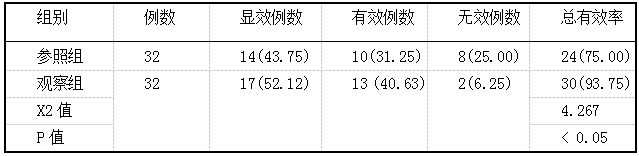 胸腺肽与恩替卡韦联用对患者原发性肝癌的临床疗效及其对预后的影响 2019.10.30 14:47
胸腺肽与恩替卡韦联用对患者原发性肝癌的临床疗效及其对预后的影响 2019.10.30 14:47 -
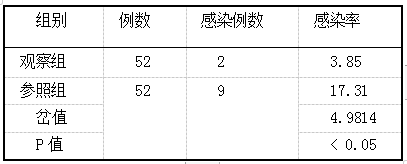 甲硝唑片官腔置入与左氧氟沙星口服对人工流产患者术后抗感染的临床疗效评价 2019.10.30 14:28
甲硝唑片官腔置入与左氧氟沙星口服对人工流产患者术后抗感染的临床疗效评价 2019.10.30 14:28 -
 瑞舒伐他汀与常规降压药物联用对反勺型高血压患者降压疗效的影响 2019.10.30 11:35
瑞舒伐他汀与常规降压药物联用对反勺型高血压患者降压疗效的影响 2019.10.30 11:35
-
 DNA的琼脂糖凝胶电泳实验原理和步骤
DNA的琼脂糖凝胶电泳实验原理和步骤 -
提取RNA及用DEPC去除RNase的实验经验
-
 医学生物学电子显微镜图谱
医学生物学电子显微镜图谱 -
 如何使用 NCBI 查找基因序列、mRNA和Promoter
如何使用 NCBI 查找基因序列、mRNA和Promoter
